Overview
SEMA (Spatial Epitope Modelling with Artificial intelligence) 2.0 is a platform to solve a number of immunology problems using artificial intelligence. SEMA provides two analyses: conformational B-cell eptiope prediction from the primary protein sequence or tertiary structure ("Predict epitopes" tab); structural comparison of antigen epitopes ("Compare epitopes" tab).
Eptiope prediction tool involves the use of sequence-based (SEMA-1D) and structure-based (SEMA-3D) approaches. SEMA-1D model is based on an ensemble of Esm-2 transformer deep neural network protein language models. SEMA-3D model is based on an ensemble of 5 pre-trained protein bimodal SaProt models. Both models were fine-tuned to predict the antigen interaction propensity of the amino acid residue with Fab regions of immunoglobulins. In addition, eptiope prediction tool comprises the model for prediction of N-glycosilation site based on the primary protein sequence. This model is based on the Esm-2 model. SEMA provides an interpretable score indicating the log-scaled expected number of contacts with antibody residues and labeled predicted N-glycosylation sites.
Epitopes comparison tool can identify structurally similar conformational epitopes between two antigens even if antigens have very low overall similarity. This tool is useful for comparison of proteins of different viral or bacterial strains and based on the neural network trained on embeddings derived from the SaProt model.
The models used in previous SEMA version accessible at GitHub.
Usage
SEMA web-platform is open access and simple to use. But for analisys of comprehensive number of sequences we recommended to use python implementaion availible on GitHub."Predict epitopes" tab
This tab allows to use conformational B-cell eptiope prediction from the primary protein sequence and tertiary structure.SEMA-1D
Input
SEMA-1D uses amino acid sequence as input data. A sequence can be submitted in two ways: paste or type a string of interest
Output
The output includes predicted epitope score and N-glycosylation label for each residue in the AA sequence. Epitope score indicates to logarithm of contact number, that is calculated as the number of antibody residues in contact with any atom of antigen residues within the distance radius of 8 Angstrom.

Visualisation of the results is color-coded protein sequence based on predicted values: brown indicates a low epitope score of zero, while cyan denotes that the epitope score exceeds the threshold (0.361), classifying the protein region as an epitope. Amino acids predicted to undergo N-glycosylation are marked with an asterisk in the sequence.
Users can click the 'Download CSV' button and download file with results in CSV format. This file contains following columns: 'Seq_pos' — position in sequense (start with 1), 'AA' — amino acid letter, 'Epitope_score' — predicted epitope score, 'N_glyco_label' — N-glycosylation label: label equal to one indictes AA undergo N-glycosylation.
SEMA-3D
Input
SEMA-3D uses tertiary structure as input data. The user can submit a tertiary structure with a target chain or an entire structure:
- type only PDB ID or PDB ID and chain(s), the corresponding structures will be extracted from pdb-file downloaded from the PDB database
- upload custom pdb-file and type a chain(s), if needed.
Output
The output includes predicted epitope score and N-glycosylation label for each residue in the AA sequence. Epitope score indicates to logarithm of contact number, that is calculated as the number of antibody residues in contact with any atom of antigen residues within the distance radius of 8 Angstrom.
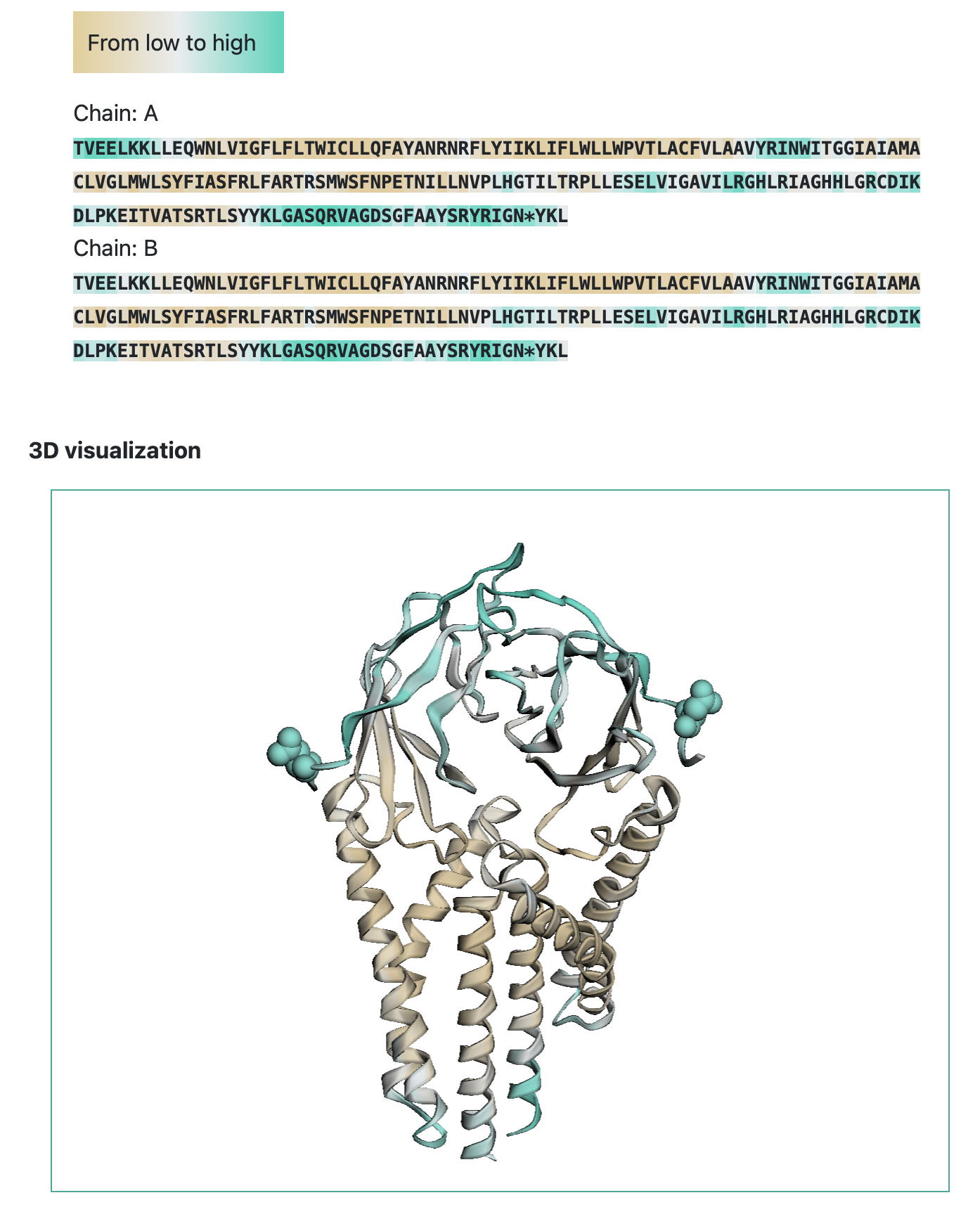
Visualisation of the results is color-coded protein sequence based on predicted values: brown indicates a low epitope score of zero, while cyan denotes that the epitope score exceeds the threshold (0.51), classifying the protein region as an epitope. Amino acids predicted to undergo N-glycosylation are marked with an asterisk in the sequence. The tertiary structure is colored as protein sequense. Amino acids predicted to undergo N-glycosylation shown as a sphere.
Users can click the 'Download results' button and download ZIP archive with results in CSV format and original PDB-file. CSV file contains prediction results presents as table with following columns: 'PDB_ID' — protein identificator (None for pdb-file submission), 'Chain' — residue chain, 'Residue position' — residue position in tertiary structure, 'AA' — amino acid letter, 'Epitope_score' — predicted epitope score, 'N_glyco_label' — N-glycosylation label: label equal to one indictes AA undergo N-glycosylation.
"Compare epitopes" tab
This tab allows to compare structural of antigen epitopes in order to find similar parts.
Input
The tool uses tertiary structures as input data. The user can submit a tertiary structure with a target chain for the proteins under study. Each structure can be submitted in two ways:
- type PDB ID and chain, the corresponding structures will be extracted from pdb-file downloaded from the PDB database
- upload custom pdb-file and type a chain.
Output
The output includes predicted similarity and epitope scores for each residue in the AA sequence for both proteins under study. Epitope score indicates to logarithm of contact number, that is calculated as the number of antibody residues in contact with any atom of antigen residues within the distance radius of 8 Angstrom. Similarity scores indicates the degree of similarity of the sites for each residue.
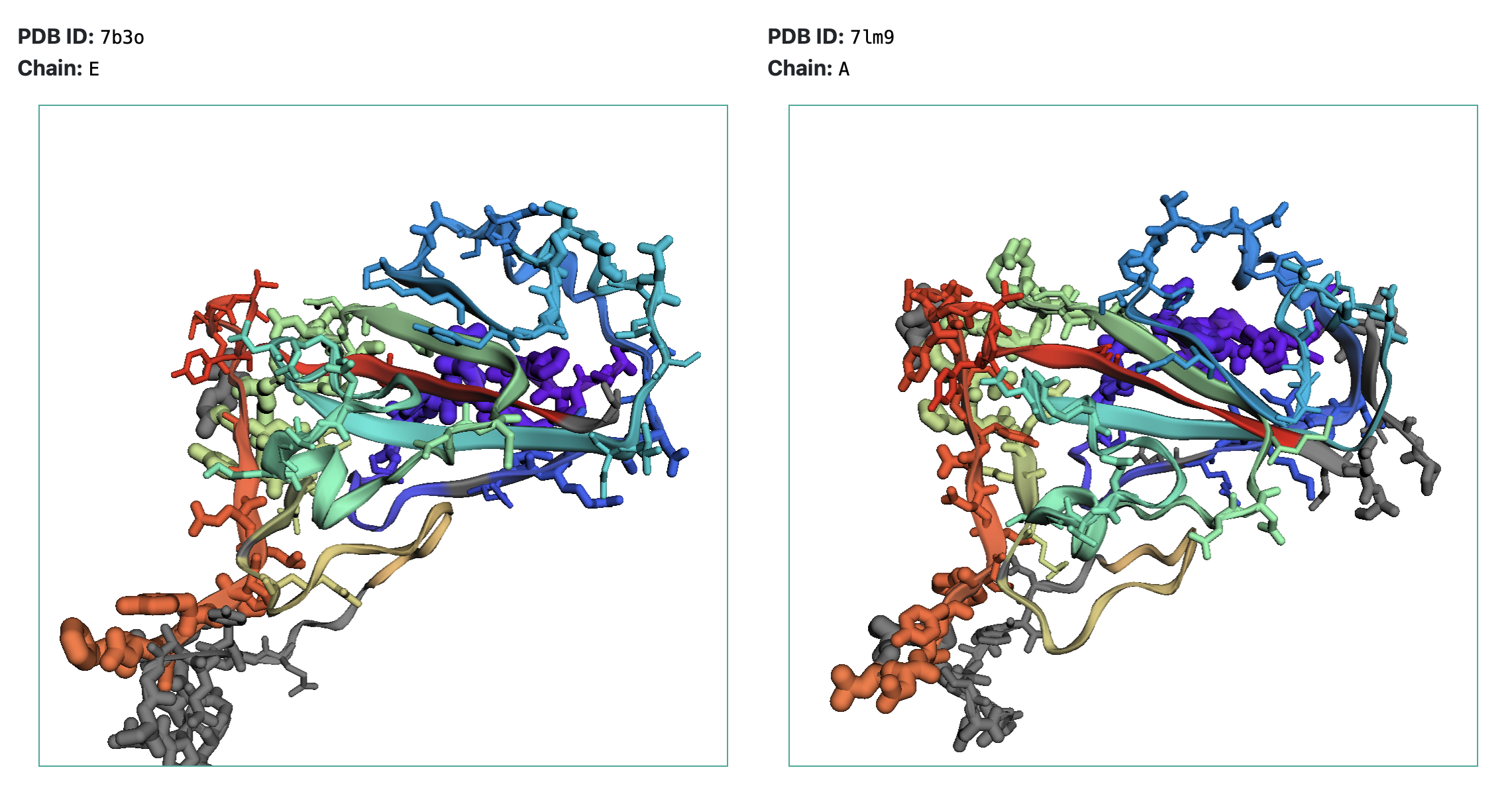
Similar parts of the teritary structures are painted in the same colors (similarity score > 2), while non-similar parts are shown in grey. Epitopes are shown as sticks, and their radius corresponds to epitope scores.
Users can click the 'Download results' button and download ZIP archive with results in CSV format and original PDB-files. Epitope prediction file, named "PDB_ID_CHAIN_SEMA2_epitopes_score.csv", contains table with following columns: 'PDB_ID' — protein identificator (None for pdb-file submission), 'Chain' — residue chain, 'Residue position' — residue position in tertiary structure, 'AA' — amino acid letter, 'Epitope_score' — predicted epitope score. Epitope comparison results contains in CSV file, named "PDB_ID1_CHAIN1_PDB_ID2_CHAIN2_SEMA2_similarity_score.csv" and presents a table with following columns: 'PDB_ID_1/2' — protein identificator (None for pdb-file submission), 'aa_1/2' — amino acid letter, 'chain_1/2' — residue chain, 'pos_1/2' — residue position in tertiary structure, 'score' — similarity score.
License
This service is free and available under MIT license.
Contuct
SEMA was developed at AIRI.
Contact e-mail: bioinformatics@airi.net
Citation
- Ivanisenko N.V., Shashkova, T.I., Shevtsov, A., Sindeeva, M., Umerenkov, D., Kardymon, O. (2024). SEMA 2.0: web-platform for B-cell conformational epitopes prediction using artificial intelligence. Nucleic Acids Research; https://doi.org/10.1093/nar/gkae386
- Shashkova, T.I., Umerenkov, D., Salnikov, M., Strashnov, P.V., Konstantinova A.V., Lebed, I., Shcherbinin, D.N., Asatryan, M.N., Kardymon, O.L., Ivanisenko, N.V. (2022). SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol.; https://doi.org/10.3389/fimmu.2022.960985